Autor: Homero Penagos G
Titulo: Tesis Para Optar Al Grado Maestria
Area:
Pais:
Perfil:
Programa:
Disponible para descarga: Yes
Ver Más Publicaciones Estudiantiles Clic aquí
Diseminar información, ideas innovadoras y conocimientos académicos es una función importante para Atlantic Internacional University. Publicaremos noticias, artículos, comentarios y otras publicaciones de nuestros estudiantes y otros colaboradores. Si desea contactar al autor por motivos profesionales favor enviar su petición por este medio.
Para conocer más de la iniciativa de Acceso Abierto de AIU haga Clic aquí.
INTRODUCCIÓN
Las enfermedades genéticas de la piel (genodermatosis) constituyen un grupo muy variado de patologías. Debido a su baja frecuencia en comparación con otras enfermedades, parecieran ser muy raras, pero consideradas en su conjunto, significan una importante contribución a las enfermedades de la edad pediátrica. (Taboada y cols., 2003)
En Panamá, un país pequeño, que tiene menos de 3 millones de habitantes (Censos Nacionales, 1990) es de esperar que su frecuencia sea aún mas baja. Tal vez por ello no hemos encontrado ninguna publicación nacional que sistematice las características de las genodermatosis en Panamá.
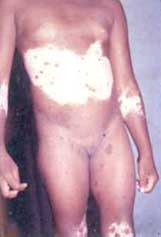
Preguntando a expertos se nos comunicó que no existe ninguna publicación al respecto (Santiago 2006, comunicación personal). En el Hospital Materno Infantil de Chiriquí, se nos menciona que desde 1995 hasta la fecha se han atendido unos 15 casos de ictiosis congénita, unos 3 casos de Síndrome de Papillón-Lefèvre, algunos casos de epiloga y frecuentes –mas de 2 al año- de Síndrome de Kindler (Dras. De León y Miranda 2006, comunicación personal). En el Hospital de niños de ciudad de Panamá, se nos dice que frecuentemente se ven Neurofibromatosis tipo I y esclerosis tuberosa, sin determinar números ( Dra. Quezada 2006, comunicación personal). Sin embargo, revisando las diferentes publicaciones médicas en Panamá desde 1930-1986 (Roy, 1988), pudimos encontrar publicaciones aisladas sobre patologías específicas (Vargas 1980, Bissot 1985, Ágreda 2004).Panamá tiene 2 poblaciones importantes afectadas por genodermatosis: los indios Kunas (San Blas y tierra firme de Colón) y los Ngöbe-Buglé que viven en las montañas de las provincias de Bocas del Toro, Chiriquí y Veraguas. En los primeros, el albinismo presenta una de las incidencias mas altas del mundo (de Oliveira Freitas, 2005) y en los segundos encontramos la mayor casuística mundial del Síndrome de Kindler (SK). (Penagos y cols., 2004)
En nuestra práctica médica hemos podido comprobar el desconocimiento sobre las genodermosis en general y el síndrome de Kindler en particular, que existe entre los profesionales de la salud que deben diagnosticar y manejar estos pacientes.
Esta tesis clasificará clínicamente las genodermatosis mas frecuentes en Panamá y actualizará el conocimiento sobre el Síndrome de Kindler. De esta forma yo deseo colaborar con la mejor atención médica de estos pacientes y proponer al gobierno nacional, la formación de una fundación para el apoyo a los pacientes afectados por estas enfermedades.
Antecedentes
Existen muchas enfermedades dermatológicas que tienen origen genético. Estas son las llamadas genodermatosis o enfermedades genéticas de la piel. Hay varias categorías de ellas clasificadas:
1.1. En relación a la afectación del material genético del afectado (a) (Abuelo, 1987)
1.1.2. En relación a la clínica expresada por el o la afectado (a) (Spitz, 2004)
En algunas situaciones hay mezcla de las clasificaciones, por ejemplo el síndrome de Kindler es un defecto de un simple gen heredado de forma autosómica recesiva, pero también es un desorden ampolloso y con fotosensibilidad. Lo importante es conocer estas 2 clasificaciones básicas, para poder saber de que estamos hablando.
Las genodermatosis particularmente, en cuanto a patologías genéticas de la piel se refiere ha aumentado en forma exponencial el conocimiento sobre ellas, pero también en cuanto a manejo y prevención de éstas (Pulkkinen, 2002).
En esta tesis usaremos la clasificación clínica modificada de Spitz (2004) por varias razones. Primero, por que creemos que el primer paso para tratar adecuadamente una genodermatosis es reconocerla y esto sólo podemos hacerlo si la diagnosticamos porque pensamos en ella y conocemos su clínica y segundo porque es nuestro deber sagrado aliviar el sufrimiento de estos pacientes, y en ella se hace énfasis en lograr esto. A continuación un breve resumen de esta clasificación.
Como se ha mencionado previamente, estas enfermedades son poco frecuentes y por ello no están todas representadas en nuestro país. En la sección de resultados y posteriormente en la de discusión ampliaremos más al respecto.
Objetivos
3.I Objetivo general
Mejorar el conocimiento de las genodermatosis en Panamá y proponer una metodología de manejo apropiada a su importancia clínica.
Pacientes, materiales y métodos
4.1. Población y muestra
Trabajo 1. Pacientes afectados de genodermatosis vistos en Chiriquí y Bocas del Toro atendidos por el autor desde 1986-2006, pacientes vistos en el Hospital Materno Infantil de Chiriquí desde 1995-2006 y pacientes del Hospital del Niño, Panamá desde 1990-2006. Es una muestra clínica por conveniencia, según fueron acudiendo a las diferentes consultas los pacientes.
Trabajo II. 26 casos atendidos en Bocas del Toro desde 1986-1996 por el autor.
Trabajo III. Pacientes revisados odontológicamente en Chiriquí Grande, 18 con SK y 13 controles.
Trabajo IV. 26 casos de SK de Panamá, 8 familiares cercanos y 12 pacientes de SK del resto del mundo.
4.2. Instrumentos de recolección de la muestra
Trabajo I. Se diseña una forma de historia clínica especialmente para recoger los datos (anexo 1).
Trabajos II, III, IV. Se obtienen los datos de artículos publicados como autor y coautor. (Ver resúmenes traducidos al español de la publicación original, anexo II).
4.3 Tipo de estudio
esta tesis se basa en 4 trabajos de investigación, 3 publicados y uno desarrollado ahora, en relación a las genodermatosis en general (Trabajo I) y al Síndrome de Kindler (Trabajos II, III, IV) en particular.
Trabajo I. Estudio descriptivo tipo transversal sobre la situación actual de las genodermatosis en Panamá.
Trabajo II. Estudio descriptivo de 26 casos nuevos de Síndrome de Kindler en Bocas del toro.
Trabajo III. Estudio de caso clínico de problemas dentales en pacientes de SK en Bocas del Toro.
Trabajo IV. Estudio clínico experimental en que se determinó el gen que causa el síndrome de Kindler.
4.4 Consideraciones éticas
Trabajos I, II, III son de tipo descriptivo, no se hace intervención, solamente se le explica al paciente lo que se va a hacer. Se les pide firmar consentimiento para tomar fotografías.
Trabajo IV. En este trabajo se realizaron medidas de tipo intervencionista como toma de muestras de sangre y biopsias de piel para microscopias de luz y electrónicas. Debido a ello fue necesario cumplir con los siguientes requisitos:
Procedimientos
Trabajo I. Los casos de genodermatosis atendidos por el autor en los Hospitales de Chiriquí y Bocas del Toro; los casos reportados como tales en el Hospital de Niños de Chiriquí y Panamá se anotarán en un documento especial para su análisis posterior.
Trabajos II, III, IV. Se revisarán los datos de Síndrome de Kindler de trabajos publicados por el autor de esta tesis. Se discutirán los resultados en la discusión general Los datos requeridos se tabularán de forma especial para este trabajo.
Resultados
5.1. Trabajo 1: Prevalencia general de genodermatosis en Panamá. Tal y como se postuló previamente solamente algunas genodermatosis fueron encontradas en Panamá (Tabla 1) de la extensa cantidad clasificadas por Spitz, 2005.
| Genodermatosis | Número | % |
Desórdenes de la formación de la queratina
|
54 |
37.5 |
Desórdenes de la pigmentación
|
22 |
15.3 |
Desórdenes mecanobulosas
|
71 |
42.4 |
Desórdenes con potencial maligno
|
2 2 |
1.4 |
Desórdenes del metabolismo de las porfirinas
|
1 1 |
0.7 |
Anormalidades cromosómicas
|
& $ |
|
| TOTAL | 150 | 100 |
* Fuente: pacientes del H. Niños de Chiriquí, Panamá y del autor. |
5.2 Trabajos ya publicados
5.2.1 Trabajo II.
Resultados
Los principales hallazgos fueron fragilidad de la piel con ampollas (100%), poiquilodermia (96%), fotosensibilidad (92%), atrofia cutánea severa (89%), hiperqueratosis de palmas y plantas (81%), Ampollas distales congénitas (81%), enfermedad periodontal severa (81%) y fimosis (80% de los pacientes masculinos). En una gran familia con 10 pacientes, el patrón hereditario fue autosómico recesivo. Tres cariotipos en pacientes y uno de un padre sano fueron normales. Los hallazgos de estudios ultraestructurales mostraron replicación de la lámina densa en 10 pacientes.
5.2.2Trabajo III
Resultados
Una gingivitis moderada a severa fue un hallazgo frecuente en todos los adultos de la población estudiada. 72% (13/18) de los pacientes de Kindler y 46% (6/13) de los controles mostraron media a severa enfermedad periodontal (p=0.001, X2). El inicio de la periodontitis fue mas precoz y la progresión mas rápida en los pacientes con Kindler. Hubo una fuerte correlación (r=0.83) entre el grado de pérdida de soporte y la edad en el grupo de Kindler y una débil correlación (r=0.66) en el grupo control). La apariencia de los tejidos gingivales sugeríaperiodontitis atípica con encías frágiles, de sangrado fácil, a veces espontáneo. En los pacientes con periodontitis, Porphyromonas gingivalis y Dialister pneumosintes fueronmas frecuentes en controles que en pacientes con Kindler
5.2.3 Trabajo IV.
Resultados
El síndrome de Kindler es una enfermedad autosómica recesiva caracterizada por ampollas neonatales, fotosensibilidad, atrofia, fragilidad y pigmentación de la piel. Estudios especiales de homocigotos de “linkage” en una cohorte aislada de pacientes y en familiares fenotípica mente sanos se mapeo en 20p12.3. Pérdida de función de mutaciones fueron identificados en el gene FLJ20116 (renombrado “KIND1”. [Encodando a Kindlin-1]. Kindlin-1 es un homólogo humano de la proteína UNC-112 de Caenorhabditis elegans, una proteína señalizante estructural asociada a la membrana que ha sido implicada en unir el cito esqueleto de actina a la matriz extracelular. Así, el síndrome de Kindler es, a nuestro conocimiento, el primer desorden de fragilidad de la piel causado por un defecto en la unión actina – matriz extracelular, mas que en la unión queratina – matriz extracelular.
Discusión
6.1 Prevalencia e importancia de las genodermatosis en Panamá
Al inicio del siglo 21 el conocimiento de las enfermedades genéticas en el ser humano ha sufrido un incremento logarítmico nunca antes visto. Esto sobre todo es debido al advenimiento de nuevas técnicas de la llamada ingeniería genética tales como:
1. La aparición de los enzimas de restricción que permiten cortar los ácidos nucleicos en puntos determinados por la propia secuencia de bases
2. La técnica de Southern que ha permitido separar, visualizar y comparar en combinación con los anterior fragmentos de ADN.
3. La técnica de la reacción en cadena de la polimerasa (PCR) que ha posibilitado acelerar y abaratar los procesos de análisis genético.
Finalmente con la reciente definición del genoma humano, la integración y la aplicación de todo este conocimiento ha llevado a la identificación de muchos genes causantes de genodermatosis a nivel mundial y también en Panamá (Siegel et al, 2003).
Teóricamente, al conocer el gen causante de la enfermedad, podría diseñarse una terapia génica para contrarrestarla, tal solución ha sido posible con la inmunodeficiencia severa (Díaz y Montiel, 2002).
Es interesante notar que al momento de conocer toda esta explosión de conocimientos sobre las genodermatosis a nivel mundial, en Panamá, aún no sabemos cuáles de ellas son mas frecuentes o prevalentes. En la Tabla I podemos ver que clínicamente encontramos desórdenes de la formación de la queratina, de la pigmentación, del tejido conectivo, mecanobulosas, con potencial maligno, del metabolismo de las porfirinas y alteraciones cromosómicas. Sin ser pocas, no son tantas como las reseñadas por Spitz (2004). Sin embargo, queremos decir que por la metodología del trabajo y por la escasa información guardada de forma estadística correcta en nuestras instituciones de salud, puede haber un subregistro importante de esta patología. Los resultados mostrados aquí pues deben ser tomados como un valor aproximado de prevalencia de 3:100,000.
A continuación, en esta sección, haremos una discusión clínica extensa de cada una de los grupos de desórdenes mas frecuentes en Panamá, toda vez, que este trabajo desea servir de guía para un mejor conocimiento de estas enfermedades. El síndrome de Kindler, dada su importancia para nosotros, se tratará en un apartado especial. Todas las fotos clínicas son de la experiencia del autor. Para el diseño de cuadros y dibujos nos basamos en el trabajo e Spitz, 2004.
6.1.1 DESÓRDENES DE LA QUERATINIZACIÓN
6.1.1.1 ICTIOSIS VULGAR (Williams, 1987; Okulicz, 2003 ; Shong, 2003; Swayder, 2004). Dibujo y figura 1.
Sinónimo
Ictiosis simple
Diagnóstico prenatal
Ninguno
Incidencia
1:250 – 1:2,000; H = M (H = hombres, M = mujeres).
Edad de presentación
Patogénesis
Hiperqueratosis por retención con proliferación epidérmica normal. Defecto en síntesis de profilagrina con su disminución intracelular en queratinocitos. Muy posiblemente una enfermedad poligénica
Guías diagnósticas
Piel: fina, leucodérmica, con escamas adherentes que respetan los pliegues y se localizan en superficies extensoras de extremidades. La cara generalmente está respetada, pero puede involucrar mejillas y frente.
Relacionada con dermatitis atópica en más de un 50% de los casos, hay queratosis pilar e hiperlinearidad palmoplantar. Rara vez hay franca queratoderma.
Diagnóstico diferencial
Dermitis atópica, xerosis, ictiosis adquirida, ictiosis ligada-X, ictiosis lamelar.
Laboratorio
Biopsia de piel afectada: ausencia de la capa granular y en microscopía electrónica se ven gránulos de queratohialina pobremente formados.
Tratamiento
Referencia a dermatología, emolientes tópicos (urea al 10%, vaselina, etc.).
Pronóstico
Mejora en clima algo frío y con la edad. Es muy variable
|
|
||||
| Dibujo1 |
|
6.1.1.2. BEBÉ COLODION (Bissot, 1985; Hennies, 1998; Huber, 2000; Larregue, 1986, Tok, 199; van gysel, 2002). Dibujo y figura 2.
Sinónimo
Ictiosis lamelar
Herencia
Autosómica recesiva, gen transglutaminasa 1 (TGM 1) en 14q11.
Diagnóstico prenatal
Amniocentesis, muestras de vello coriónico. Estudios de mutación del gen o de linkage en familias donde el
Defecto molecular es conocido. Biopsia de piel fetal a las 22 semanas.
Incidencia
Menos de 1:300,000; H = M.
Edad de presentación
Al nacimiento.
Patogénesis
Mutaciones heterogéneas en el gene TGM 1 interfiere con el normal entrecruzamiento de las proteínas estructurales en la capa lípida y proteica de la epidermis superior llevando a una deficiente queratinización y descamación.
Guías diagnósticas
Piel: En el recién nacido vemos una membrana translucente en todo el cuerpo, ectropium, eclabium, eritroderma generalizada con riesgo de sepsis secundaria y deshidratación hipernatrémica. La membrana se descama a los primeros días o semanas de vida. En el niño y adulto hay grandes escamas, brillantes en los pliegues, eritrodermia, ectropium, queratodermia palmoplantar y disminución de la sudoración con intolerancia al calor.
Pelo: alopecia cicatricial.
Uñas: distrofia secundaria con inflamación del pliegue ungueal.
Diagnóstico diferencial
Hiperqueratosis epidermolítica, ictiosis ligada-X, eritroderma ictiosiforme congénita, síndrome de Netherton, tricotiodistrofia.
Laboratorio
Biopsia de piel para demostración in situ de expresión y actividad de TGM 1. Si hay alopecia examen de pelo con microscopio de luz. En el recién nacido estudio por sepsis.
Tratamiento
Recién nacido: manejo en una unidad de cuidados intensivos neonatal, manejo de líquidos, electrolitos; manejo de sepsis. Uso de emolientes. Uso de cámara de hidratación.
Niño / adulto: Retinoides, emolientes, evite ejercicios extenuantes, mucho calor y de consejo genético.
Pronóstico
Sufrimiento a lo largo de toda la vida, pero con expectativas normales de larga vida.
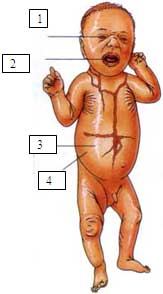
|
|
||||
Diagrama 2. Bebé colodión |
|
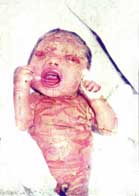
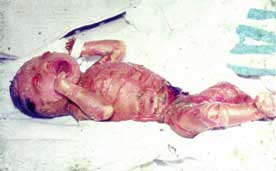
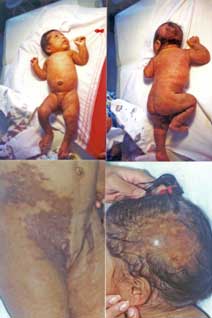
6.1.1.3 ICTIOSIS ARLEQUÍN (Vargas, 1980; Bianca, 2003; Chan, 2003; Dale, 1990; Michel, 1999). Dibujo y figura 3.
Sinónimo
Ictiosis arlequín
Herencia
Mas posiblemente autosómica recesiva, heterogenecidad genética con descripción reciente de nueva delección en 18q21.
Diagnóstico prenatal
Amniocentesis: células de fluido amniótico con morfología anormal. Ultrasonido. En biopsia de piel fetal ausencia de cuerpos lamelares en la microscopía electrónica.
Incidencia
Menos de 1: 300,000; H = M.
Edad de presentación
Al nacimiento
Patogénesis
Se han descrito diversas alteraciones genéticas y moleculares. Todos los pacientes tienen lo siguiente en común: defectos de queratinización con los queratinocitos con anormal diferenciación morfológica y bioquímica que lleva a un exceso de hiperqueratosis; un error del metabolismo lípido que lleva a acumulo de éstos en el estrato córneo; ausencia de gránulos normales lamelares; deficiente conversión de profilagrina a filagrina y una disminución de calpaina ( una proteasa activada por calcio importante en la señalización mediada por calcio para la diferenciación celular) que podría jugar un papel en el fenotipo; hay 3 subtipos descritos en base a la expresión de diferentes proteínas queratinas, presencia de profilagrinas y presencia y número de gránulos lamelares.
Guías diagnósticas
Piel: Placas masivas de hiperqueratosis encasillando la piel del recién nacido. Ectropión severo; eclabium; pabellones auriculares, nariz, dedos de pies y manos deformes o ausentes; pobre regulación del calor corporal. Descamación generalizada con eritrodermia en los supervivientes del período neonatal.
Diagnóstico diferencial
Eritrodermia ictiosiforme congénita severa, ictiosis lamelar severa.
Laboratorio
Estudio por sepsis
Tratamiento
En una unidad de cuidados intensivos neonatal con manejo de fluidos y electrolitos. Para sepsis antibióticos sistémicos; incubadoras humidificadas. El uso de retinoides puede desprender la escama y mejorar la probabilidad de sobrevivir. Si sobrevive más allá de neonato debe referirse al cirujano para corrección de orejas, nariz, dedos o al dermatólogo para uso de retinoides y emolientes. Al oftalmólogo para ectropión y queratitis
Pronóstico
Si no muere al nacer, ésta viene por problemas respiratorios o intestinales por sepsis o por la obstrucción retrictiva. Reportes de sobrevivencia con uso de retinoides han sido publicados.
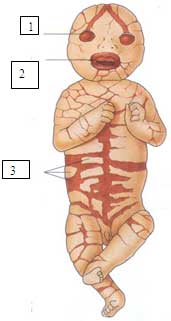
|
|
||||
Dibujo 3. Ictiosis arlequín |
|
6.1.1.4 QUERATODERMIA PALMO PLANTAR DIFUSA (QPPD)
(Devos, 2003; Drechsler, 1994). Figura 4.
Sinónimo
QPPD de Vörner, epidermolítica, no-epidermolítica, Unna-Thost.
Herencia
Autosómica dominante; Vörner : gen de queratina 9 (la mas común) y 1(17q21 y 17q13 respectivamente)Unna-Thost: gen de queratina 1.
Diagnóstico prenatal
Análisis de linkage y detección de mutación.
Incidencia
1.200 a 1:40,000, H = M.
Edad de presentación
Luego del nacimiento y hasta un año después.
Patogénesis
La mutación de los genes de la queratina 1 y 9 daña el ensamble de los filamentos de queratina dentro de la piel palmo-plantar.
Guías diagnósticas
Piel: hiperqueratosis simétrica difusa palmar y plantar con un tinte blanco-amarillento; bien demarcadas con borde eritematoso; no se extiende al dorso; fisuras dolorosas y puede haber dermatofitosis. Puede haber hiperhidrosis y/o bromhidrosis. En la variedad Unna-Thost puede haber extensión al dorso de manos y pies. Marcha anormal por el dolor.
Diagnóstico diferencial
Todas las otras formas de QPPD
Laboratorio
Biopsia de piel: variante Unna-Thost: ortoqueratosis; variante Vörner: hiperqueratosis epidermolítica.
Tratamiento
Referencia al dermatólogo (emolientes, queratolíticos, retinoides orales, antimicóticos, PUVA terapia. Referencia al podólogo (debridar, emoliente).
Pronóstico
Persiste toda la vida con una esperanza de vida normal.
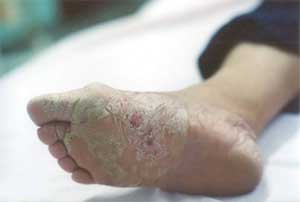
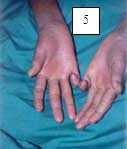
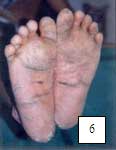
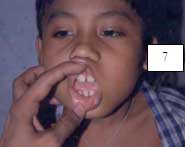
6.1.1.5 SÍNDROME DE PAPILLON- LEFÈVRE (Hart, 1998; Allende, 2003; Almuned, 2003; Laass, 1997; Ullbro, 2003). Figuras 5, 6, 7.
Sinónimo
Queratoderma palmo plantar con periodontosis
Herencia
Autosómica recesiva; CTSC gen en 11q14
Diagnóstico prenatal
No
Incidencia
Mas de 120 casos reportados.
Edad de presentación
Desde el nacimiento a los 5 años de edad.
Patogénesis
Mutaciones en el gen CTSC que codifica la catapepsina C, que es una proteasa lisosomal.
Guías diagnósticas
Piel: Queratoderma palmoplantar claramente demarcada con borde eritematoso y puede extenderse al dorso y al tendón de aquiles, hiperhidrosis, mal olor y queratoderma puntata.
Cabello: puede ser escaso
Boca: periodontitis con severa gingivitis; resorción del hueso alveolar; pérdida de dientes deciduos y permanentes.
Sistema nervioso central: calcificación dural del tentorium y del anclaje coroideo.
Diagnóstico diferencial
Otras formas de QPPD
Laboratorio
Radiografías dentales, resonancia magnética de cerebro, cultivos por bacterias.
Tratamiento
Referencia al odontólogo (higiene oral meticulosa, remoción regular de la placa, antibióticos específicos, dentaduras postizas.
Referencia al dermatólogo (queratolíticos tópicos, antibióticos orales, retinoides).
Pronóstico
La periodontitis se resuelve cuando todos los diente se han caído; la pérdida prematura de los dientes puede llevar a distorsión del crecimiento de los huesos mandibulares y maxilares. La queratodermia persiste a través de toda la vida.
|
|
|||||||||
 |
 |
|||||||||
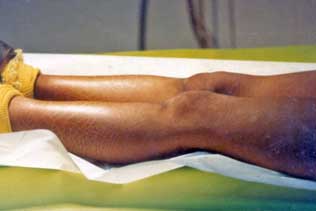
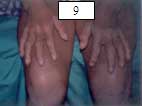
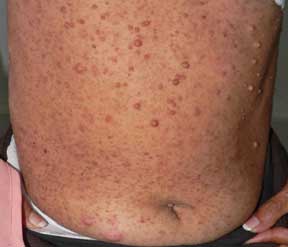
| Fig. 8 hiperqueratosis en codos. | Fig. 9 Hiperqueratosis en dorso de manos y rodillas. Aunque estos son diversos pacientes; todos los hallazgos clínicos son frecuentes en un mismo paciente. | |||||||||
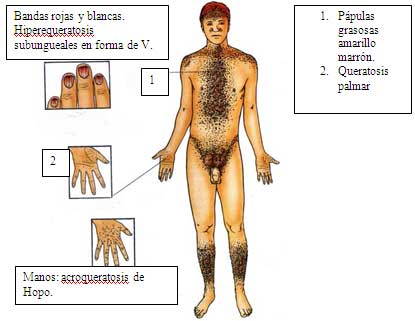
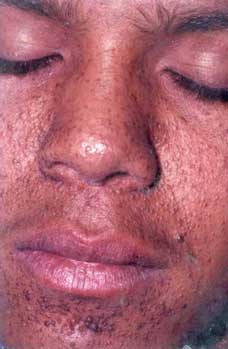
6.1.1.6 ENFERMEDAD DE DARIER (Bashir, 1993; Burge, 1992; Hulatt, 2003). Dibujo 4, figuras 10, 11,12.
Sinónimo
Enfermedad de Darier–White; queratosis foliculares.
Herencia
Autosómica dominante; gen ATP2 A2 en 12q23-24.
Diagnóstico prenatal
Analisis de linkage de DNA.
Incidencia
1:55,000 – 1:100,000; H = M.
Edad de presentación
La segunda década de vida, rara vez en la edad adulta.
Patogénesis
Mutaciones en el gen ATP2 A2 que codifica SERCA2 (sarcoendoplasmic reticulum
Ca2+ ATPase isoform 2) una bomba de calcio que aumenta la extracción intracelular de éste para el normal diferenciamiento y desarrollo epidérmico.
Guías diagnósticas
Piel: pápulas hipequeratósicas que coalescen en forma verrucosa en distribución seborreica en cabeza, cara, tronco, pliegues e inguinales; son amarillas a chocolates, conmal olor y grasosas. Pueden ser infectados por herpes simple virus o por bacterias. Tienen fotosensibilidad UVB. Tienen placas verrugosas en dorso de manos (llamada acroqueratosis verruciforme de Hopo) y queratolisis puntata en palmas y plantas.
Uñas: bandas rojas y blancas alternas longitudinales; hiperqueratosis subungueales; fracturas en V en placa distal y con o sin roturas de la placa ungueal.
Membranas mucosas: pápulas en empedrado en mucosas de boca y ano genital.
Sistema nervioso central: Esquizofrenia y retardo mental se han reportado en algunas familias.
Diagnóstico diferencial
Enfermedad de Hailey – Hailey; enfermedad de Grover; pitiriasis rubra pilaris; psoriasis.
Laboratorio
Biopsia de piel.
Tratamiento
Referencia a dermatología (retinoides sistémicos, tratamientos tópicos, etc.)
Pronóstico
Crónicos empeorando en invierno; duración de vida normal.
|
|
||||||
 |
 |
||||||
| Fig. 11 Pápulas hiquerqueratósicas en un paciente blanco en donde se pueden ver en área de espalda superior, dichas lesiones marrón grasientas. | Fig. 12 Queratosis puntata en planta de paciente que le molestan a la deambulación. |
6.1.2 DESÓRDENES DE LA PIGMENTACIÓN
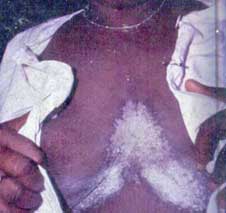
6.1.2.1 ALBINISMO OCULOCUTÁNEO TIPO 1 (de Figueriedo, 2005, Bassi, 1995) (Fig. 13)
Sinónimo
Albinismo tirosinasa positivo (tipo 2, el mas común)
Herencia
Autosómico recesivo gen P en 15q11.2-12
Diagnóstico prenatal
Análisis de linkage y de detección de mutación.
Incidencia
1:15,000 negros; 1:37,000 blancos, 1:100 indios Kunas de San Blas, Panamá, H = M.
Edad de presentación
Al nacimiento
Patogénesis
La mutación del gen P que disminuye la síntesis de eumelanina; el producto de este gen se cree juega un papel en el transporte de tirosinasa; (+) tirosinasa, número normal de melanocitos, disminución de melanina piel, cabello y ojos. Hay mal distribución de las fibras ópticas.
Guías diagnósticas
Piel: color crema a rosado; nevos pigmentados múltiples, pecas y léntigenes aumentan con la edad; queratosis solar y cánceres baso celulares o escamosos de la piel con la edad.
Cabello: color crema a amarillo chocolate.
Ojos: iris azul a amarillo – marrón (dependiente de la raza); nistagmos; fotofobia; disminución de la agudeza visual; estrabismo; hipoplasia de la fovea.
Diagnóstico diferencial
Otros tipos de albinismo; síndrome de Hermansky–Pudlak; síndrome de Chédiak-Higashi
Tratamiento
Piel: evitar el sol (en especial al medio día); uso de protectores solares; protección con ropas. Referencia a dermatología para vigilar cáncer de piel cada 6 meses.
Ojos: lentes con vidrios con filtros ultravioletas; lentes correctivos; referencia a oftalmología.
Pronóstico
Puede mejorar en general con la edad.

Fig. 13 Niño albino de la etnia Kuna, Panamá. Observe el cabello blanco, la piel rosada-blanca que contrasta con
la piel del familiar que lo carga. Observe la desviación de los ojos, que ya vislumbra problemas oculares frecuentes en ellos.
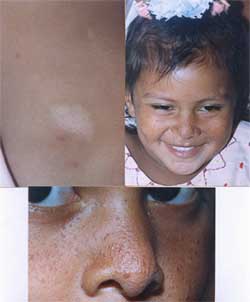
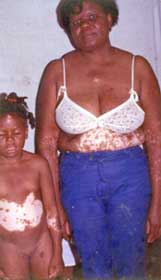
6.1.2.2 PIEBALDISMO O ALBINISMO PARCIAL (Butttazzoni, 2005; Thomas, 2004) Figs. 14, 15.
Sinónimo
Manchas blancas familiares
Herencia
Autosómica dominante por mutación de c-kit proto-oncogéno 4q12.
Diagnóstico prenatal
Análisis de linkage de DNA y de detección de mutación.
Incidencia
Menos de 1:20,000; todas las razas; M = H.
Edad de presentación
Al nacimiento.
Patogénesis
Anormalidad en los receptores transmembranas tirosinasa kinasas; disminuyendo la señal de transcripción; anormalidad en la embriogénesis de los melanocitos con proliferación, migración y distribución defectiva de los melanoblastos
Diagnóstico diferencial
Síndrome de Waardenburg; viitiligo, devus despigmentado.
Laboratorio
En biopsia de áreas depigmentada hay ausencia o disminución de melanocitos y de melanina.
Tratamiento
Injertos antólogos de melanocitos, protectores solares, maquillaje corrector, monobencil éter de hidroquinona como depigmentante.
Pronóstico
Expectativa de vida normal y discromía estable y permanente (a menos que se trate).
 |
|
|||
Fig. 14 Madre e hijas afectadas (autósomica recesiva). Observe el mechón blanco en la niña y lesiones de hiperpigmentación dentro de las leucodérmicas. |
|
6.1.2.3 INCONTINENCIA PIGMENTARIA (Bross, 2005; Aggarwal, 2003) Figs. 17,
Sinónimo
Síndrome de Bloch-Sulzberger
Herencia
Dominante ligada-X; raro que un hombre afectado sobreviva, a menos que sea un Klinefelter. NEMO gen en Xq28.
Diagnóstico prenatal
Análisis de DNA si el gen es conocido en la familia.
Incidencia
Unos 700 casos reportados, 97 % mujeres.
Edad de presentación
Al nacimiento o a las primeras semanas de vida.
Guías diagnósticas
Piel
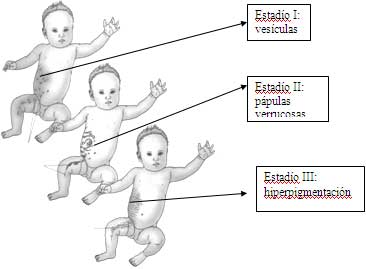
Estadío 1- Vesicular: nacimiento-1-2 semanas de vida, vesículas y bulas en patrón linear en extremidades, tronco y piel cabelluda. Máculas y pápulas eritematosas.
Estadío 2 – Verrucosa: 2-6 semanas. Líneas de pápulas y pústulas hiperqueratosis en extremidades.
Estadío 3 – Hiperpigmentada: 3-6 meses. Remolinos y ondas de hiperpigmentación siguiendo las líneas de Blaschko.
Estadío 4 – Hipopigmentada: 2-3 décadas de vida. Hipopigmentación en reemplazo de la anterior hiperpigmentación con/sin atrofia folicular.
Cabello: alopecia cicatricial en 30%.
Uñas: cambios distróficos en 5-10 de los casos.
Dientes: afecta los deciduos y permanentes, adontia y en un 66% dientes cónicos o en clavijas.
Ojos: estrabismo, catarata, atrofia óptica, cambios vasculares retinales con ceguera secundaria y posibles masas retrolentales, todo esto en un 25-30%
Sistema nervioso central: convulsiones, retardo mental y parálisis fláccida hasta en un 30%.
Diagnóstico diferencial
Epidermolisis bulosa, impétigo, herpes simple virus, hiperqueratosis epidermolítica, sífilis
congénita, hipomelanosis de Ito.
Laboratorio
En vesicular abundante eosinófilos; eosinofilia en la infancia en hemograma.
Tratamiento
Dermatología (diagnóstico, cuidado local); odontología(al año de vida); oftalmología al momento del diagnóstico; neurología si hay sintomatología.
Pronóstico
Esperanza de vida normal
|
|
||||||||||||
 |
 |
||||||||||||
| Dibujo 6 | Fig. 17 Madre de la niña de foto anterior, solo hipopigmentación, ninguna de las otras características presenta. |
||||||||||||
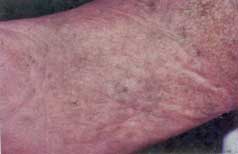
6.1.2.4 SÍNDROME DE ALBRIGHT (Lee, 1986; De Sanctis, 1999; Ringel, 1996) Fig. 17.
Sinónimo
Síndrome de McCune - Albright
Herencia
Esporádico; mutación somática post - cigótica en el gen CNAS1 en 20q13.2
Diagnóstico prenatal
Ninguno
Incidencia
Raro; H = M.
Edad de presentación
Al nacer o en los primeros meses de vida.
Patogénesis
Mutaciones por mosaicismo en el gen CNAS1 que codifica la unidad α para la proteína G estimuladora que regula la adenil ciclasa.
Guías diagnósticas
Piel: manchas café con leche grande, festoneada, como la forma de la costa de Maine.
Hueso: displasia fibrosa poliostótica afectando huesos largo y faciales, a veces debajo de la
Mancha café con leche. Esclerosis difusa en la base del cráneo, fracturas recurrentes, acortamiento de extremidades.
Endocrino: pubertad precoz, mujeres más que hombres. Hipertiroidismo (20-30% de los casos.
Diagnóstico diferencial
Neurofibromatosis múltiple 1.
Laboratorio
Rayos X, fosfatasa alcalina sérica (elevada). Pruebas tiroideas. En las mujeres LH, estrógenos, 17-OH-corticoides, 17b cetoesteroides.
Tratamiento
Ortopedia (corrección quirúrgicas), endocrinología
Pronóstico
Esperanza de vida normal
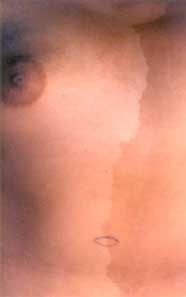
Fig. 18. Paciente indígena, con 11 años, menarca a los 10 años y desarrollo incipiente de caracteres sexuales.
Con esta lesión de nacimiento. Sitio marcado de biopsia de piel. Gran mancha café con leche en todo tórax y abdomen anterior izquierdo.
6.1.2.5 NEUROFIBROMATOSIS TIPO 1 (Barker, 1987) Dibujo 7; Figs. 19, 20.
Sinónimo
Enfermedad de von Ricklinghausen; NF - 1
Herencia
Autosómica dominante; gen locus 17q11.2; mutación espontánea en el 50% de los casos.
Diagnóstico prenatal
Análisis de mutación de DNA si el gen es conocido.
Incidencia
1:3,000; todas las razas; H = M. 85% de todos los casos, la forma mas frecuente.
Edad de presentación
Al nacimiento o temprano en la infancia.
Patogénesis
Disminución o ausencia de neurofibromina (producto del gen), es un supresor de tumores que regula la producción de ras proto-oncogénos lo que puede llegar a progresión del tumor cuando ocurre la mutación.
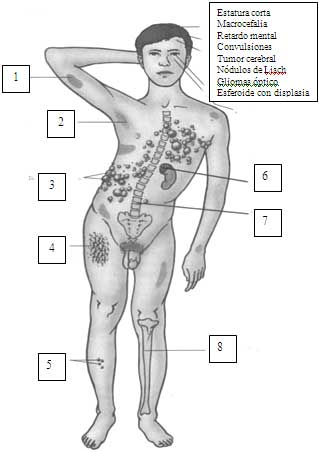
Guías diagnósticas
Piel (ver también criterios diagnósticos)
Mácula café con leche (aumento en número y tamaño en los primeros 5 años de vida). Más de 6 sugieren NF-1.
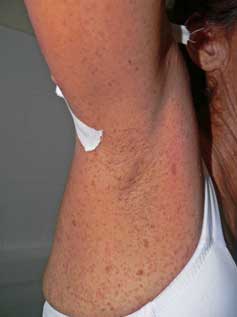
Pecas en axilas o inguinal
Neurofibromas:dérmicos o subcutáneos, aparecen en la pubertad, aumentan en número con edad y el embarazo, frecuentes en tronco. Hay congénitos plexiformes, de crecimiento progresivo, con o sin hiperpigmentación, hipertricosis. Prurito puede sentirse sobre un neurofibroma en crecimiento.
Ojos (ver criterios diagnósticos): nódulos de Lisch en el iris en hamartomas (vistos en más de 60% pacientes mayores de 5 años), aumentan en número con la edad, son asintomáticos. Glaucoma congénito y nevo coroideo.
Neoplasia
Sistema nervioso central: glioma óptico (66% asintomático) puede causar ceguera si no se trata. Otros como astrocitomas, meningiomas, chuanoma vestibular (neuroma acústico), ependimoma.
Sistema esquelético (ver criterios diagnósticos)
Displasia alar del esfenoides, macrocefalia, escoliosis (sobre todo cervico-torácica), displasisa de discos vertebrales, seudoartrosis de tibia, estatura corta.
Sistema Nervioso Central
Convulsiones, defectos para hablar e incoordinación inespecífica, hidrocéfalo, cefalea.
Vascular
Displasia vascular con compromiso cerebral, gastrointestinal y renal; infartos cerebral; hipertensión renovascular.
Diagnóstico diferencial
Neurofibromatosis 2; otras formas de neurofibromatosis; síndrome de McCune Albright; síndrome de Watson (una variante alélica); síndrome de Noonan (puede ocurrir simultáneamente); síndrome Proteus.
Criterios de consenso diagnóstico de los Institutos Nacionales de la Salud, 1987.
Laboratorio
Resonancia magnética de cráneo y de columna vertebral de inicio (control)
Tratamiento
Historia completa y examen físico total.
Examen oftalmológico basal
Control regular con pediatra, internista, oftalmólogo, neurólogo, dermatólogo.
Referencia a ortopedia, audiología, psiquiatría, cirugía, neurocirugía, oncología, si es necesario.
Examinar los familiares en primer grado de descendencia.
Pronóstico
Si padece un neurofibrosarcoma, feocromocitoma, complicaciones vasculares o en el
SNC. La variabilidad en severidad de la enfermedad puede ser muy grande.
|
|
|||||||
 |
||||||||
| Fig. 20 Igual paciente con más neurofibromas de diferentes tamaños. Por lo demás, totalmente sana. | ||||||||
6.1.2.6 ESCLEROSIS TUBEROSA (Bader, 2003; Dabora, 2001; Imán, 2000; von Slegtenhorst, 1999)
Sinónimo
Síndrome de Bourneville; epiloia.
Herencia
Autosómica dominante por gen TSC1 en 9q34 o TSC2 en 16p13. Mas del 66% es por mutación espontánea.
Diagnóstico prenatal
Ecocardiografía fetal mostrando un rabdomioma; análisis e DNA si la mutación es conocida.
Incidencia
1:10,000; H = M; todas las razas.
Edad de presentación
Al nacimiento
Guías diagnósticas
Piel
Máculas hipopigmentadas: en hojas de fresno: hallazgo precoz, muy característico; poligonales: mas comunes; en confeti: en piel pretibial.
“Piel de sapa” nevo de tejido conectivo.
Angiofibromas faciales: llamados “adenomas sebáceos”.
Fibromas periungueales.
Placas fibrosas en cara.
Mácula café con leche.
Sistema nervioso Central
Espasmos infantiles, convulsiones tónico-clónicas, hipsarritmia, retardo mental, tumores corticales, calcificación para ventriculares, nódulos subependimales con o sin hidrocéfalo obstructivo, astrocitomas.
Ojos: hamartomas retinales (facomas).
Riñones: angiomiolipomas, quistes.
Cardiacos: rabdomioma.
Oral: fibromas encías, fragmentación del esmalte.
Músculo esquelético: quistes falanges, engrosamiento periostio.
Pulmón: linfangiomatosis.
Diagnóstico diferencial
Nevo depigmentoso, Hipomelanosis gutata idiomática, nevo anémico, vitiligo.
Laboratorio
Examen con luz de Word; ultrasonido transfontanela; resonancia magnética, electroencefalograma, examen de fondo de ojo, ultrasonido renal, ecocardiografía en la infancia.
Tratamiento
Examen físico completo y seguimiento por su médico de cabecera. Referencia a quien corresponda (neurología, neurocirugía, etc).
Valoración cuidadosa cutánea y general a familiares de primer grado de consanguinidad.
Pronóstico
Muerte prematura por complicaciones cardiovasculres
|
|
||||||
 |
 |
||||||
| Fig. 22 Angiofibromas en cara de paciente de 21 años con convulsiones. | Fig. 23 Mácula hipocrómica en flanco de abdomen, en forma de |
6.1.3 DESÓRDENES MECANO BULOSAS
6.1.4.1 EPIDERMOLISIS BULOSA SIMPLE (Epstein, 1992; Fine, 1991;Fontao, 2004)
Sinónimo
Ninguno
Herencia
Autosómica dominante; muy rara vez autosómica recesiva; en gen queratina 5 sobre 12q y en gen queratina 14 sobre 17q.
Diagnóstico prenatal
Análisis de DNA.
Incidencia
10-30 casos por millón de nacimientos vivos; H = M.
Edad de presentación
Weber-Cockayne = 1-3 década de vida
Koebner (generalizada)= nacimiento a temprana infancia.
Dowling-Meara= nacimiento a primer mes de vida.
Patogénesis
Las mutaciones ya mencionadas producen un cito esqueleto basilar débil por daños a los filamentos intermedios de queratina con fragilidad mecánica resultando en bulas intraepidérmicas después de trauma. Mutaciones del gene plectina afectan la proteína hemidesmosómica y juega un papel en la EB con distrofia muscular y la variante Orna.
Guías diagnósticas
Weber-Cockayne (WC)
Piel: bulas palmoplantares, callos, hiperqueratosis; dolorosas o no, con infección o no, empeora en veranos o clima caliente.
Generalizada o Koebner (K)
Piel: bulas generalizadas con o sin infección; empeora en veranos o clima caliente.
Boca: erosión leve de mucosa.
Dowling-Meara: (DM)
Piel: bulas diseminadas con distribución herpetiforme, puede ser mortal. Con discromía postinflamatoria, no cicatricial, produce milia y con la edad desarrollan queratodermia palmoplantar.
Uñas: distrofia con posible pérdida.
Membranas mucosas: ampollas, erosiones en boca o esófago.
Diagnóstico diferencial
WC: paquioniquia congénita, tiña pedis, eccema dishidrótico, sífilis congénita.
K y DM: herpes simple virus neonatal, sepsis por bacterias, incontinencia del pigmento, sífilis congénita, impétigo bulosa, enfermedad por Ig A linear.
Laboratorio
Bulas intraepidérmica en biopsia de piel, en microscopia electrónica se ven tonofilamentos gruesos en DM; inmunomapeo con anticuerpos monoclonales. Cultivos bacteriales y virales. Análisis de DNA con muestras de mucosa oral y sangre.
Tratamiento
Referencia a dermatología = diagnóstico, evitar trauma, cuidados de la herida, manejo en general del paciente.
Referencia a podiatría = suelas ortopédicas de silicona o plastizoato, uso de medias delgadas blancas de algodón, para evitar roce y sudoración excesiva. Si hay muchas bulas, manejar en unidad de cuidados intensivos neonatales, con manejo de fluidos, electrolitos y antibióticos, si es necesario.
Pronóstico
Esperanza de vida normal, pero debilitada. Las ampollas tienden a disminuir con la edad. Con DM puede haber mortalidad en los primeros meses de vida.
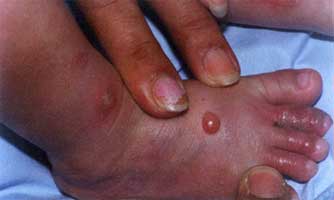
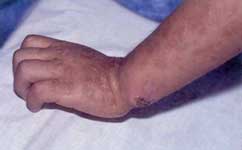
Fig. 24 Paciente con 3 meses de edad, ampollas cerradas y rotas en pies y manos.
6.1.4 DESÓRDENES CON POTENCIAL MALIGNO (Poole, 1993)
6.1.4.1 XERODERMA PIGMENTOSO
Herencia
Autosómico recesivo
Gen DDB1 en 9q22, XPA gen codifica proteína 1 unión para daño al DNA
Gen ERCC3 en 2q21, XPB gen codifica el complemento cruzado 3 de excidir y reparar.
XPC gen que codifica endonucleasas en 3p25.
XPD gen que codifica ERCC2 en 19q13.
XPE gen que codifica DDB2 en 11p12
XPF gen que codifica Ercc4 en 16p13
XPG gen que codifica endonucleasa en 13q33.
XPV (variante) gen que codifica a η polimerasa en 6p21.
Diagnóstico prenatal
Análisis de DNA. Ensayo de síntesis no esquematizada de DNA sobre células de fluido amniótico cultivadas.
Incidencia
1:1,000,000 en Estados Unidos de América; 1:40,000 en Japón, H = M.
Edad de presentación
Primeros pocos años de vida.
Patogénesis
Las mutaciones en los genes que codifican enzimas de reparación del DNA (helicasa, endonucleasa; o la proteína de unión al daño de DNA) llevan a un proceso defectuoso en la escisión – reparación del DNA cuando es expuesta a la radiación ultravioleta. Puede haber deficiente reparación post replicación en el grupo XPV.
Guías diagnósticas
Infancia: fotosensibilidad extrema con quemaduras solares.
Skin (la severidad varía con muchos grupos complementarias)
Primer año de vida
Niñez-adolescencia (listada en orden de aparición): máculas pigmentadas, máculas acrómicas, máculas y telangectasias en foto distribución. Piel seca, atrófica y descamativa; con estrechamiento de los orificios nasales y oral. Riesgo aumentado de queratosis, queratoacantoma; riesgo 1,000 aumentado de cáncer de piel de todos los tipos.
Ojos
Síntomas progresivos de fotofobia, conjuntivitis, telangectasias y pigmentación de de párpados y conjuntiva, ectropión, vascularización corneal, opacidad de córnea, papilomas benignos en párpados; carcinoma basal y melanoma.
Neurológicos (20% de los casos, mas en XPA y XPD.
Daño progresivo con retraso mental, sordera sensorineural, microcefalia, hiporeflexia, espasticidad, ataxia.
Diagnóstico diferencial
Infancia y niñez temprana: porfiria eritropoyética, porfiria congénita eritropoyética, Síndrome de Bloom etc.
Laboratorio
Análisis de DNA. Ensayo no ordenado de DNA con cultivo de fibroblastos cutáneos.
Tratamiento
Referencia a dermatología = evitar el sol, protección física y química. Lentes a prueba de sol y usar cabellos largos. Vigilar por cáncer de piel cada 3 meses y cada semana por familiar entrenado por el médico. Remoción precoz de queratosis, lesiones precancerosas y cáncer de p’iel.
Referencia a oftalmología = usos de lágrimas artificiales (carboximetil celulosa), lentes blandos, transplante de córnea, vigilar por cáncer.
Referencia a neurología si es necesario.
Referencia a grupos de apoyo de XP.
Pronóstico
Diagnóstico precoz y adecuado manejo, regular pronóstico; de lo contrario mueren temprano. Los daños neurológicos evolucionan sin tener que ver con la radiación ultravioleta. Causa severo daño psico social.
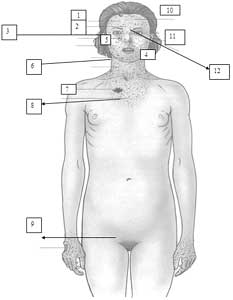
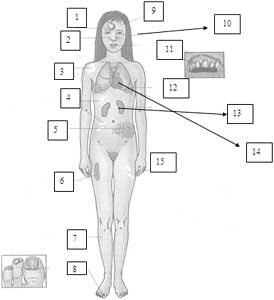
Dibujo 9. 1. Queratosis actínicas; 2. Telangectasias; 3. Carcinoma basal; 4. Atrofia despigmentada; 5. Queratoacantoma; 6. Máculas hiperpigmentadas; 7. Melanoma; 8. Fotodermatitis; 9. Carcinoma escamosos; 10. Retardo mental, hiperreflexia, espastiicidad y ataxia; 11. Sordera sensorineural; 12. Ectropión, opacidad corneal, papilomas palpebrales, fotofobia, conjuntivitis, melanoma uveal.
6.1.5 DESÓRDENES DEL METABOLISMO DE LAS PORFIRINAS
6.1.5.1 PORFIRIA CONGÉNITA ERITROPOYÉTICA (PCE) (Dawe, 2002; Xu, 1996)
Sinónimo
Enfermedad de Günter.
Herencia
Autosómica recesiva; gen UROGEN III en 10q25.2-q26 (gen uroporfirinógeno sintetaza).
Diagnóstico prenatal
Análisis de DNA; disminución de la actividad de la enzima en cultivo de células amnióticas.
Incidencia
Menos de 200 casos reportados, es muy rara.
Edad de presentación
En la infancia a niñez temprana.
Patogénesis
Las mutaciones de gen de la sintetaza de UROGEN III producen fenotipo.
Guías diagnósticas
Piel
Tempranas: inmediata fotosensibilidad con quemaduras, edema, eritema vesículas y bulas, erosiones, infección.
Tardías: escaras mutilantes con deformidad de la nariz, oídos y dedos; alopecia cicatricial; discromías; cambios esclerodermiformes.
Pelo: hipertricosis tipo lanugo en cara, cuello y extremidades.
Ojos: fotofobia, ectropium, conjuntivitis.
Dientes: coloración rojo o violeta de dientes deciduos y permanentes; fluorescencia rojo coral con luz de Wood.
Hematología: anemia hemolítica.
Diagnóstico diferencial
Porfiria hepatoeritropoyética, otras porfirias, epidermolisis bulosa, xeroderma pigmentoso, penfigoide buloso.
Laboratorio
Análisis de enzima con glóbulos rojos: nivel de uroporfirinógeno III sintetaza disminuido; determinar el nivel de porfirinas plasmáticas y el espectro de fluorescencia; aumento notable de urorporfirina en glóbulos rojos, plasma y orina; aumento de coproporfirinas en excremento.
Tratamiento
Referencia a dermatología
Evitar el sol, protectores solares físicos, químicos, ropas que le cubran bien. Antibióticos para la infección. B carotenos, para efecto fotoprotector.
Referencia a cirugía: esplenectomia para mejorar la anemia hemolítica.
Transfusiones de glóbulos rojos empacados, hematina intravenosa, carbón activado todas con algo de utilidad y limitados por efectos secundarios.
Referencia a odontología: coronas de porcelana o acrílico.
Pronóstico
Si es bien llevado y manejado muy buena esperanza de vida; si no la calidad de vida puede verse muy afectada.
|
|
|||||||
 |
||||||||
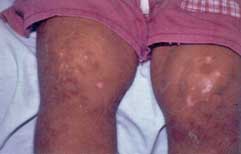
Fig. 27 Ampollas, cicatrices, hiperpigmentación en rodillas. |
||||||||
6.1.6 ANORMALIDADES CROMOSÓMICAS
6.1.6.1 SÍNDROME DE DOWN (Bittles, 2004; Hayes, 1993; Schepis, 1994; Scherbenske, 1990).
Sinónimo
Síndrome de la Trisomía 21
Herencia
2% de los casos asociados a una translocación heredada, de otra forma no es heredado.
Diagnóstico prenatal
Análisis de cromosomas por medio de estudio de líquido amniótico demuestra trisomía 21.
Ultrasonido en 2/3 trimestre: una constelación de anormalidades puede sugerir el diagnóstico: hidrops fetal, higroma quístico, defectos cardíacos, edema de nuca, anormalidad en abdomen en ciruela (arrugado), obstrucción duodenal.
Incidencia
Aproximadamente 1:700; M = H; 45% de los afectados con madre mayores de 45 años. 1% de recurrencia en parientes de niños afectados por no disyunción; 50% de aborto espontáneo en segundo trimestre.
Edad de presentación
Al nacimiento
Patogénesis
95% de los casos secundarios a no disyunción del cromosoma 21 durante la meiosis de uno de los padres (95% de las madres). De un 4-5 % se deben a una translocación (heredada de novo). El resto 1-2% se debe a mosaicismo que ocurre como un evento postcigótico.
Guías diagnósticas
Piel
Surco palmar único (palma de simio), pezones planos, aumento de los pliegues de la nuca en la infancia, siringomas, elastosis perforante serpiginosa, xerosis y liquenificación con la edad, propensión a las infecciones.
Cabello: alopecia areata.
Craneofacial: braquicefalia, cara plana, puente nasal aplanado con nariz pequeña, occipucio plano, cuello ancho y corto, oídos pequeños con pabellones auriculares displásicos o ausentes.
Ojos: pliegues epicantales, fisuras palpebrales horizontales, manchas de Brushfield, opacidades finas del cristalino, estrabismo.
Boca: pequeña con lengua escrotal protruyente, labios fisurados y gruesos, anormalidades dentales, enfermedad periodontal.
Músculo-esquelético: estatura corta, hipotonía en la infancia, manos pequeñas y anchas por acortamiento de falanges y metacarpos; clinodactilia del 5 dedo; espacio aumentado entre el 1 y 2 dedo de los pies. Anormalidades odontoideas con inestabilidad del eje Atlanta-axial. Crestas iliacas planas con ángulo estrecho acetabular
Sistema Nerviosos Central: retardo mental (IQ = 30-50); convulsiones (10%)
Cardiovascular: enfermedad congénita (AV comunicación y defecto septo ventricular)
Gastrointestinal: atresia duodenal es muy común.
Hematológicos: leucemia mielógena agua, en el recién nacido reacción leucemoide pasajera y policitemia; inmunodeficiencia.
Endocrinos: hiper o hipotiroidismo autoinmune.
Genitourinario: pene pequeño, disminución de la líbido en el varón, mas frecuente la impotencia.
Diagnóstico diferencial
Ninguno.
Laboratorio
Análisis cromosómico, Rayos X de pelvis, pruebas funcionales de tiroides, conteo hematológico completo, ecocardiograma.
Tratamiento
Completo seguimiento de su médico de atención primaria y referencia oportuna al menor síntoma. Dado que los problemas cardíacos pueden ser mortales precozmente, algunos centros hacen cirugía cardiaca muy tempranamente.
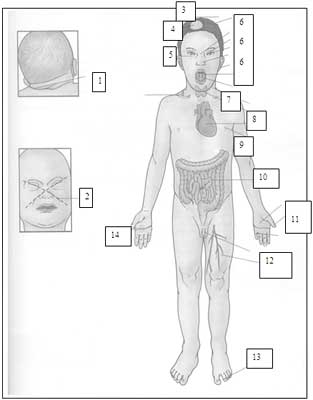
Dibujo 10. 1. Pliegues redundantes en la nuca y cuello corto; 2. Signo de la X al llorar el bebé; 3. Estatura corta; 4. Alopecia areata; 5. Siringomas; 6. Retardo mental, convulsiones, puente nasal planos, pliegues epicánticos, manchas de Brushfield, anormalidades dentales, nariz pequeña, oídos displásicos, enfermedad periodontal, macroglosia, lengua escrotal; 7. Hipotiroidismo; 8. Malformaciones cardíacas; 9. Pezones planos; 10. Anomalías gastrointestinales; 11. Manos anchas, clinodactilia; 12. Pene pequeño, leucemia mielógena aguda; 13. Separación entre 1 y segundo dedo; 14. Pliegue único palmar.
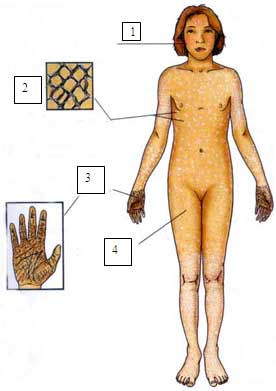
6.2 Situación actual del síndrome de Kindler en Panamá
6.2.1 SÍNDROME DE KINDLER (Bruyneel, 1996; Patrizi, 1996; Penagos, 2004; Siegel, Ashton, Penagos y cols. 2003; Wiebe, Penagos; 2003; Freiman, 2005)
Sinónimo
Poiquilodermia congénita bulosa con atrofia cutánea.
Herencia
Autosómica recesiva; KIND 1 gen (codifica Kindlin-1) en 20p12.3. Hay casos esporádicos comunes sobre todo en familias con consanguinidad. Expresión variable del gen entre familias también ha sido reportada.
Diagnóstico prenatal
No
Incidencia
Menos de 100 casos reportados desde 1954 a 2002. En Panamá 20:100,000 (Siegel, Ashton, Penagos y cols., 2003.
Edad de presentación
Al nacimiento o a los primeros meses de vida.
Patogénesis
El gen involucrado tiene que ver con la correcta formación de la matriz extracelular del citoesqueleto de actina. El SK sería la primera genodermatosis causada por un defecto como el mencionado, diferenciándose finalmente de otros síndromes de fragilidad cutánea.
Guías diagnósticas
Diagnóstico diferencial
Síndrome de Bloom, Síndrome de Cockayne, Disqueratosis congénita, epidermolisis bulosa, eritroqueratodermia varaiabilis, Síndrome de Rothmund-Thomson, Xeroderma Pigmentoso.
Laboratorio
Análisis de mutación del gen KIND 1. Histología de luz de la piel atrófica revela cambios de poiquiloderma (epidermis aplanada y atrófica, edema en unión dermoepidérmica, vacuolización focal con degeneración de las células basales. Hay prominencia de capilares dérmicos, incontinencia pigmentaria, infiltrado linfocítico perivascular). Disminución de la tinción con anticuerpos anti-kindlin-1 de posibles pacientes comparados con controles.
La microscopia electrónica muestra reduplicación y disrupción de la lámina densa con fibrillas de anclaje a lo largo de la unión dermoepidérmica, sugiriendo continua remodelación de la zona de la membrana basal. En bulas hay 3 niveles de daño: intraepidérmicas, dentro de la lámina lúcida (de unión), por debajo de la lámina densa (dérmica).
Tratamiento
Médico: sintomático y preventivo, evitar el trauma, evitar el sol y usar fotoprotección. Antibióticos y cuidado de heridas si hay.
Quirúrgico: corrección de estenosis uretrales, anales o esofágicas.
Referencia: dermatología, genetista, dentista, psicólogo, oftalmología o cirugía.
Pronóstico
Bueno para la esperanza de vida. En nuestros pacientes indígenas, sin cuidado médico adecuado –hasta el momento- por lo distante y aislados que viven, complicaciones como infecciones pueden causar mortalidad en la infancia.
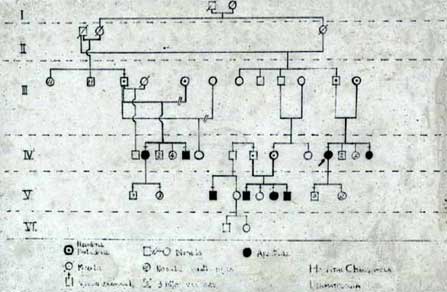
Fig. 28 Patrón de herencia de una familia que cubre 6 generaciones en donde se observa un patrón de herencia autosómica recesiva y se puede ver claramente como los padres portadores tienen hijos enfermos.
Leyenda:
Cuadros = varones
Círculos = mujeres
Cuadros o círculos con un punto = portadores del gen enfermo
Círculos o cuadros negros = enfermos, tienen el gen recesivo doble
Círculos o cuadros con raya diagonal = fallecidos
Círculos o cuadros con una cruz en su parte superior = que han sido examinados por quien realizó el cuadro de historia familiar.
|
|
||||||
 |
 |
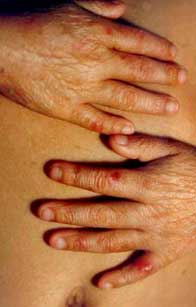
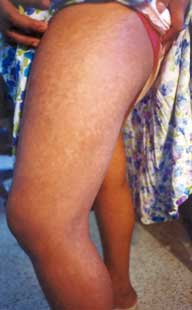
||||||
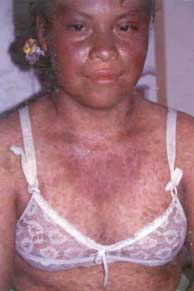
Fig. 31 Atrofia, discromía (hiper-hipo pigmentación) con |
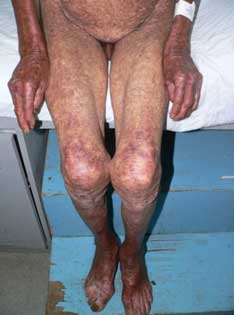
Fig. 32 Poiquilodermia severa en un paciente de 19 años. Este un |
||||||
 |
 |
||||||
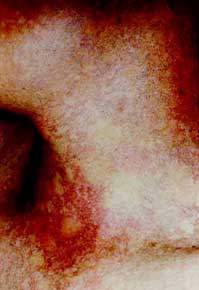
Fig. 33 Atrofia severa de la piel en un niño de 7 años. |
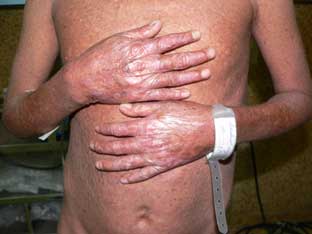
Fig. 34 Atrofia epidérmica universal de un paciente de 80 años. |
||||||
 |
 |
||||||
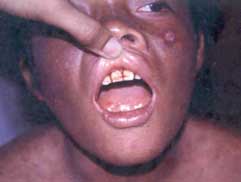
Fig. 35. Gingivitis hemorrágica, piorrea y daño del hueso alveolar en un paciente de 17 años |
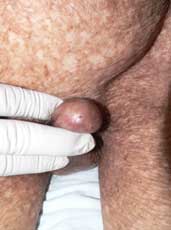
Fig. 36 Fimosis preesente en el 80% de los pacientes de Panamá, un hallazgo clínico no descrito antes. Como profilaxis nosotros recomendamos circuncisión en todos los nuevos pacientes de Síndrome de Kindler diagnosticados. Este es un paciente de 80 años. Note la discromía y atrofia universal que presenta. |
||||||
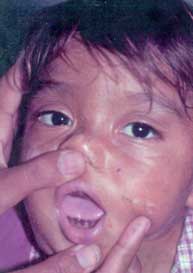
 |
 |
||||||
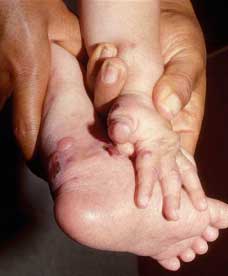
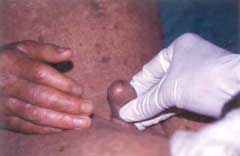
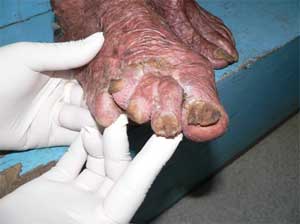
Fig. 37 Fimosis en un paciente de 5 años. Note las mismas |
Fig. 38 La fusión de dedos ha sido descrito en Kindler, en este paciente de 80 años podemos verte este fenómeno llamado también sindactilia en su pie derecho, en el izquierdo era similar. Las manos eran normales. De nuestros 66 casos, hemos encontrado solamente unos 6 (9%). |
||||||
 |
|||||||
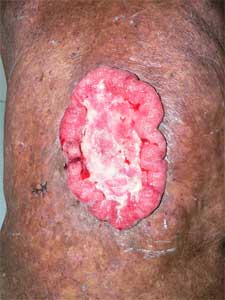
Fig. 39 Se han descrito carcinomas de piel y de vejiga. Este gran carcinoma escamoso de la espalda es el único caso que hemos detectado en nuestros 66 pacientes. Este es un paciente de 80 con 5 años de evolución en la montaña de esta lesión. La remoción del tumor involucró hasta muy cerca de la columna vertebral. |
|||||||
Conclusiones Y Recomendaciones
Las genodermatosis comprenden una fracción no frecuente dentro de las enfermedades en general, pero si causan morbilidad importante en la población pediátrica y en sus familias. Una genodermatosis importante es el síndrome de Kindler y afecta población vulnerable, muy pobre que no tiene acceso a un adecuado cuidado de salud.
Recomendación: diseñar un plan integral de manejo de estas patologías, el cual adjuntamos en el anexo 1.
| Abuelo D. | Genetic principles. Dermatologics Clinics 1987; 5:1=15. |
| Aggarwal K, Jain VK, Dayal s | Incontinentia pigmenti with nail dystrophy. Dermatol Venereol Leprol 2003; 69:7; pp. 3-4. |
| Almuneef M, AKS, AAA et al. | Pyogenic liver abscess and Papillon-Lefevre sydrome not a rare association. Pediatrics 2003;111:e85-e88. |
| Allende LM, Moreno AU, | A genetic study of cathepsin C gene in two families with Pillon-Lefevre syndrome. Mol Genet Metab 2003;79:146-148. |
| Bader RS, Chitayat D, Kelly E, et al. | Fetal rhabdomyoma: prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex. J Pediatr, 2003;143:620-624. |
| Barker D, Wright E, Nguyen K, et al | Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 1987 May 29; 236(4805): 1100-2[Medline]. |
| Barzilai DA, Freiman a, Dellavalle RP et al | Dermatoepidemiology. J am Acad Dermatol 2005; 52: 559-73. |
| Bashir R, Munro CS, Mason S, et al. | Localisation of a gene for Darier´s disease. Human Mol Genet1993;2:1937-1939. |
| Bassi MT, Schiaffino MV, Renieri A, et al | Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet 1995 May; 10(1): 13-9 [Medline]. |
| Bianca S, Ingegnosi C, Bonaffini F. | Harlequin foetus. J Posgrad Med 2003:49:81-82 |
| Bissot AA, Herrera D, Poveda R. | Ictiosis Lamelar. Revista del Hospital del Niño, 11:6; 1985. |
| Bittles AH, Glasson EJ. | Clinical, social, and ethical implications of changing life expectancy in Down síndrome. Dev Med Child Neurol 2004;46:282-286. |
| Bross SM | Incontinencia pigmenti accesado en https://imgen.bcm.tmc.edu/IPIF |
| Bruyneel-Rapp F, Mallory SB, Luke MC. | What Syndrome is this. Pediatric Dermatology, 1996. |
| Burge SM, Wilkinson JD | Darier-White disease : a review of clinical features in 163 patients. J Am Med Dermatol 1992;27:40-50. |
| Buttazzoni S, Pale J, Schrön R y cols | Piebaldismo (Albinismo parcial). Hoja Hospitalaria 3. 2005, 2:3, pp. 29-31. |
| Contraloría de la República | Censos Nacionales de Panamá, 1990. |
| Chan YC, Tay YK, Tan LK, et al. | Arlequín Ictiosis in association with hypothyroidism and juvenile rheumatoid arthritis.Pediatr Dermatol 2003; 20:421-426. |
| Dabora SL, Joswiak S, Franz DN, et al | Mutational analysis in a cohorte of 224 Tuberous Sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organ. Am J Hum Genet 2001;68:64-80. |
| Dale BA, Holbrook KA, Fleckman P, et al. | Heterogeneity in harlequin ichthyosis, an inborn error of epidermal queratinization: variable morphology and structural protein expression and a defect in lamellar granules. J Invest Dermatol 1990; 94:6-18. |
| Dawe Sa, Peters TJ, Du Vivier A, et al | Congenital erythropoieitic porphyria: dilemmas in present day management. Clin Exp Dermatol 27:680-683. |
| Drappier JC, Khosrotehrani K, Zeller J, et al. | Medical management of neurofibromataosis I: a cross-sectional study of 383 paatients. J Am Acad Dermatol 2003;49:440-444 |
| De Oliveira FF, Figueredo FJ, Alves SJ | Albinismo en comunidades indígenas (O fator cultural afetando a prevalencia de doenca). Fundacao Nacional de Saude, Brasil, 2005. |
| De Sanctis D, Lala R, Matarazzo P, et al. | McCune-Albright syndrome: a longiitudinal clinical study of 32 patients. J Pediatr Endocrnoli Metab 1999;12:817-826. |
| Devos SA, Daleschuse J. | An unusual case of palmoplantar keraatoderma. J Eur Acad Dermatol Veneorol 2003;17:68-69. |
| Díaz GM, Montiel G. | Terapia génica en inmunodeficiencia primarias. Rev Med Hosp. Nac Niños (Costa Rica), v. 37 n. 1-2 (accesado el 07/11/2005 en https://www.scielo.sa.cr/scielo.php?script=sci_arttex&pid=S1017-85462002000100008&) |
| Drechsler M, SchrockE, Royer-Pokora B | Keratin 9 gene mutations in epidermolytic palmoplantar keratoderma. (EPPK) Nat Genet 1994;6:174-179. |
| Epstein EH Jr. | Molecular genetics of epidermolysis bullosa. Science 1992;256:799-804. |
| Fine JD, Bauer EA, Briggaman RA, et al | Revised clinical and laboratory criteria for subtypes of inherited epidermolysis bulosa: a consensus report by the subcommittee on diagnosis and classification of the National Epidermolysis Bullosa Registry. J Am Acad Dermatol 1991;24:119-135. |
| Fontao L, Tasanen, K, Huber M, et al | Molecular consequences of deletion of citoplasmic domain of bullous pemphigoid 180 in a patient witn predominant features of epidermolyis bullosa simplex. J Invest Dermatol 2004;122:65-72. |
| Freiman A, Russell L. | Kindler Syndrome, eMedicine electrónica accesado en https://emedicine.com.com/derm/topic943.htm el 6/11/05 |
| Hart TC, Bowden DW, Ghaffar KA, et al | Sublocalization of the Papillon-Lefevre syndrome locus on 11q14-q21. Am J Med Genet 1998 Sep 1; 79(2): 134-9[Medline]. |
| Hayes A, Batshaw ML | Down syndrome.Ped Clin North Am 1993;40:523-535. |
| Hennies HC, Kuster W, Wiebe V, et al. | Genotype(phenotype correlation in autosomal recessive lamellar ichthyosis . am Hum Genet 1998; 62:1052-1061 |
| Huber M, Limat A, Wagner E, et al. | Efficient in vitro transfection of human keratinocytes with an adenovirus-enhanced receptor-mediated system. J Invest Dermatol 2000,; 114:661-666. |
| Hulatt L, Burge S. | Darier’s disease: hopes and challenges. J R Soc Med 2003;96:439-445. |
| Myman MH, Whittemore VH | National Institus of Health Consensus Conference: tuberous sclerosis complex. Arch Neurol 2000; 57:662-665. |
| Kluwe L, Siebert R, Gesk S, et al. | Screeninig 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat 2004;23:111-116. |
| Larregue M, Ottavy N, Bressieux JM, et al. | Collodium baby: 32 new cases reports. Ann Derm Venereol 1986; 113:773-785. |
| Laass MW, Hennies HC, Preis S, et al. | Localization of a gen for Papillon-Lefevre to chromosome 11q14-q21 by homozygosity mapping. Human Genet 1997; 101:376-382. |
| Lee PA, Van Dop C, Migeon CAJ. | McCune-Albright syndrome: long-term follow-up. JAMA 1986;256:2980-2984. |
| Micali G, Bene-Bain MA, Guitart J, Solomon LM | Genodermatoses; In: Pediatric Dermatology, Schachner LA ed, Second Edition, Churchill Livinstone, New York, 1995, pp 347-468. |
| Michel M, Fleckman P, Smith IT, et al. | The calcium-activated neutral protease calàin I is present in normal foetal skin and is decreased in neonatal harlequin ichthyosis. Br J Dermatol 1999; 141:1017-1026. |
| Okulicz JF, Chwartz Ra. | Hereditary and acquired ichthyosis vulgaris. Int J Dermatol 2003;42:95-98. |
| Patrizi A, Pauluzzi P, Neri I et al. | Kindler Syndrome: report of a case with ultrastructural study and review of the literature. Pediatric Dermatology 1996; Vol 13 No. 5; 397-402. |
| Penagos H, Jaen M, Sancho MT et al. | Kindler Síndrome in native americans from Panama. Arch Dermatol 2004; 140:939-944. |
| Poole S, Fenske NA | Cutaneous markers of internal malignancy. I. Malignant involvement of the skin and the genodermatoses. J AM Acad Dermatol 1993;28:1-13. |
| Pulkkinen L, Ringpfeil F, Uitto J. | Progress in heritable skin diseases: molecular bases and clinical implications. J Am Acad Dermatol 2002; 47: 91-104. |
| Roy, A | Historia y análisis de las revistas médicas en Psnsm´s, II parte 1977-1980. Rev Med CSS 1981;13:445-626. |
| Schepis D, Barone C, Siragusa,M et al | An updated surveyon skin conditions in Down syndrome. Dermatology 2002;205:234-238. |
| Scherbenske JM, Benson PM, Rotchford JP, et al | Cutaneous and ocular manifestations of Down syndrome. J Am Acad Dermatol 1990;22:933-938. |
| Sellheyer K, Belbin TJ. | DNA microarrays: From structural genomics to functional genomic. The applications of gene chips in dermatology and dermatopathology. J Am Acad Dermatol 2004; 51: 681-92. |
| Shwayder T | Disorders of keratinization: dianosis and management. Am J Clin Dermatol 2004;5:17-29. |
| Siegel DH, Ashton GHS, Penagos HA et al. | Loss of Kindlin-1, a human homologo f the Caenorhabditis elegans actin-extracellular-matriz linker protein UNC-112, causes Kindler Síndrome. Am Hum Genet 2003; 73:74-187. |
| Spitz, JL | Genodermatosis, a clinical guide to genetic skin disorders. 2004, Lippincott Williams Wilkins, Phil, 2nd edition. |
| Taboada N, Lardoeyt R. | Criterios para el diagnóstico clínico de algunos síndromes genéticos. Rev Cubana Pediatr v.75 n.1 2003[citado 09 Mayo 2006], p.0-0. Disponible en la World Wide Web: <https://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312003000100007&lng=es&nrm=iso>. ISSN 0034-7531. |
| Thomas I, Kihiczak GG, Fox MD, et al | : Piebaldism: an update. Int J Dermatol 2004; Oct; 43(10): 716-9[Medline]. |
| Tok J, Garzon MC, Cserhalmi-Friedman P, et al. | Identification of mutations in the transglutaminase 1 gene in lamellar ichthyosis. Exp Dermatol 1999; 8:128-133. |
| Ullbro C, Crossner CG, Nederfors T, et al. | Dermatologic and oral finfings in a cohort of 47 patients with Papillon-Lefevre syndrome. J Am Acad Dermatol 2003;48:345-351. |
| Van Gysel D, Lijnen RL, Moekti SS, et al. | Collodion baby: a follow-up study of 17 cases. J Eur Acad Dermatol Venereol 2002; 16:472-475. |
| Van Slegtenhorst M, de Hoogt R, Hermans, C, et al | Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997; 277:805-808. |
| Vargas PE, Carrera E y cols. | El feto arlequín: una dermatosis ictiosiforme letal. Boletín de la Sociedad Panameña de Pediatría, IX, 18; 1980. |
| Xu W, Astrin KH, Desnick RJ, et al | Molecular basis of congenital erythropoieitic porphyria muttions in the human uroporphyrinogen III synthetase gen. Hum Mutat 1996;7:187-192. |
| Wiebe CB, Penagos H, Luong N y cols | Clinical and microbiologic study of periodontitis associated with Kindler Syndrome. Journal of Periodontology, 2003, 74 (1), pp. 25-31. |
| Williams ML, Elias PM | Genetically transmitted, generalized disorders of cornifiation. The Ichthyosis. Dermatol clin 1987;5:155-178. |
| Zhong W, Cui B, Zhang Y, et al. | Linkage analysis suggests a locus of ichthyosis vulgaris on 1q22. J Hum Genet2003;48:390-392. |
Anexos
ANEXO I
RESÚMENES TRADUCIDOS AL ESPAÑOL DE LOS ARTÍCULOS PUBLICADOS.
ARTICULO II
Síndrome de Kindler en indígenas de Panamá. Reporte de 26 casos.
Homero Penagos, MD; Marta Jaén, MD; Mario T. Sancho, MD; Manuel R. Saborío, MD; Víctor G. Fallas, MD; Dawn H. Siegel, MD; Ilona J. Frieden, MD
Objetivo: Investigar las características clínicas, genéticas y de laboratorio de 26 casos de síndrome de Kindler.
Diseño: Los pacientes fueron captados cuando acudían a la consulta externa de dermatología en el Hospital de Changuinola, Bocas del Toro, Panamá durante mayo de 1986 a diciembre de 1990.
Procedimientos: La historia clínica, examen físico y estudios de laboratorio fueron hechos en un hospital comunitario en Panamá. A doce de los pacientes se les hicieron exámenes hechos en el Hospital de Niños de Costa Rica.
Pacientes: Un total de 26 pacientes participaron en el estudio. Ellos eran miembros de la etnia Ngöbe-Buglé y Vivian en zonas aisladas rurales. Panamá.
Resultados: Los principales hallazgos fueron fragilidad de la piel con ampollas (100%), poiquilodermia (96%), fotosensibilidad (92%), atrofia cutánea severa (89%), hiperqueratosis de palmas y plantas (81%), Ampollas distales congénitas (81%), enfermedad periodontal severa (81%) y fimosis (80% de los pacientes masculinos). En una gran familia con 10 pacientes, el patrón hereditario fue autosómico recesivo. Tres cariotipos en pacientes y uno de un padre sano fueron normales. Los hallazgos de estudios ultraestructurales mostraron replicación de la lámina densa en 10 pacientes.
Conclusiones: Para nuestro conocimiento este estudio representa la mayor serie de pacientes con síndrome de Kindler hasta la fecha. Las características clínicas confirman casos reportados y los análisis de segregación confirman el tipo de herencia como autosómica recesiva. Nosotros reportamos también la fimosis como una complicación, la cual no había sido reportada previamente en este síndrome.Arch Dermatol. 2004;140:939-944
ARTÍCULO III
Estudio clínico y microbiológico de la periodontitis asociada al síndrome de Kindler.
. Journal of Periodontology 2003, Vol. 74, No. 1, Pages 25-31
Dr. Colin B. Wiebe , Department of Oral Biological & Medical Sciences, Faculty of Dentistry, University of British Columbia, Vancouver, BC. Homero Penagos , Department of Dermatology, Hospital Region of Chiriqui, Social Security Bureau of Panama, David, Panama. Nancy Luong, University of Southern California School of Dentistry, Los Angeles, CA. Jørgen Slots, University of Southern California School of Dentistry, Los Angeles, CA. Ervin Epstein Jr., Department of Dermatology, San Francisco General Hospital Medical Center, University of California, School of Medicine, San Francisco, CA. Dawn Siegel, Department of Dermatology, San Francisco General Hospital Medical Center, University of California, School of Medicine, San Francisco, CA. Lari Häkkinen, Department of Oral Biological & Medical Sciences, Faculty of Dentistry, University of British Columbia, Vancouver, BC. Edward E. Putnins,, Department of Oral Biological & Medical Sciences, Faculty of Dentistry, University of British Columbia, Vancouver, BC. Hannu S. Larjava , Department of Oral Biological & Medical Sciences, Faculty of Dentistry, University of British Columbia, Vancouver, BC.
Antecedentes: Poco es conocido acerca del inicio y Prevalencia de la enfermedad periodontal en pacientes con el raro síndrome de Kindler que es un desorden tipo genodermatosis. Este estudio investiga el nivel de unión periodontal clínica en relación a la edad y presencia de posibles bacterias atogenéticas en individuos con síndrome de Kindler.
Artículo IV
Pérdida de Kindlin-1, un homólogo humano de Caenorhabditis elegans, proteína UNC-112 fijadora de la matriz extracelular de Actina, causa el síndrome de Kindler.

El síndrome de Kindler es una enfermedad autosómica recesiva caracterizada por ampollas neonatales, fotosensibilidad, atrofia, fragilidad y pigmentación de la piel. Estudios especiales de homocigotos de “linkage” en una cohorte aislada de pacientes y en familiares fenotipicamente sanos se mapeo en 20p12.3. Pérdida de función de mutaciones fueron identificados en el gene FLJ20116 (renombrado “KIND1”. [encodando a Kindlin-1]. Kindlin-1 es un homólogo humano de la proteína UNC-112 de Caenorhabditis elegans, una proteína señalizante estructural asociada a la membrana que ha sido implicada en unir el citoesqueleto de actina a la matriz extracelular. Asi, el síndrome de Kindleres, a nuestro conocimiento, el primer desorden de fragilidad de la piel causado por un defecto en la unión actina – matriz extracelular, mas que en la unión queratina – matriz extrracelular.
ANEXO II
FUNDACION PARA EL MANEJO Y PREVENCIÓN DE LAS GENODERMATOSIS EN PANAMÁ
PROPUESTA
DR. HOMERO PENAGOS G.
DERMATÓLOGO, CSS PANAMÁ
CHIRIQUÍ, MAYO 2006
RESÚMEN EJECUTIVO
Fundación
La Fundación es una organización voluntaria, sin fines de lucro, dedicada a promover la investigación y la educación sobre las Genodermatosis en Panamá y otras enfermedades severas de la piel, además de proporcionar tratamientos y apoyo a los pacientes y a sus familias.
Estamos dedicados a mejorar la calidad de vida de los pacientes con G, y otras enfermedades severas de la piel y de sus familias.
Los propósitos de la Fundación son:
Promover y apoyar la investigación científica para estudiar las causas y el tratamiento de la G y de otras enfermedades genéticas de la piel.
Aliviar el sufrimiento físico y mental de las personas con G a través de consejos prácticos, guía y soporte.Obtener información acerca de la enfermedad, y proporcionarlos a los pacientes con G y a sus familias.
Ayudar a las personas con G a encontrar atención médica, social y genética de acuerdo a sus necesidades.
Actuar como fuente de información para los médicos y otros profesionales de la salud.
Distribuir material informativo sobre la G al público en general.
Presidente :
Vice-Presidente:
Tesorera (o):
Vocales:
Director Ejecutivo:
Coordinadora Médica:
Secretaria:
Contador:
Domicilio de la Fundación:
Chiriquí
Dirección de la Fundación:
Calle B Sur
Dirección Postal :. Apdo 855
Teléfonos 774 15 03Fax: 774 15 03
Coreo Electrónico:
Pagina Web: www.genodermatosispanama.com
Personería Jurídica:
Cuentas de banco:
Objetivo de la Fundación:
La fundación tiene como objetivo promover y apoyar la prevención, investigación, las causas, los tratamientos y buscar la cura mas frecuentes en Panamá para así ofrecer una mejor calidad de vida a los niños que padecen las mencionadas enfermedades y a sus familiares.
Miembros Fundadores:
Dr. Homero Penagos
Dra. Tirza de León
Dr. Gherson Cukier
Prof. Elvia Requena de Penagos
Dra. Marta Jaén de Oliveros
Lcdo. Jorge Requena
MEd Ruth Requena
Prof. Elba González
Dr. Marino Rivera
Dr,. Frankin Anguizola
Dr. Felipe Villarreal
Dr. Abel Gómez
Lcdo. Felipe Rodríguez
Lcdo. Fernando Mendizábal
Lcda. Estrellita Fernández
Miembros Honorarios
Vivian Fernández de Torrijos Su Excelencia Primera Dama de Panamá
Asesores Médicos extranjeros:
Ervin Epstein Jr, MD
INTRODUCCIÓN
TIPOS DE GENODERMATOSIS MÁS FRECUENTES EN PANAMÁ
Tal y como hemos podido ver en el trabajo anterior, las genodermatosis mas frecuentes en Panamá son: el albinismo óculo cutáneo tipo I, el síndrome de Kindler, Neurofibromatosis tipo I y el Síndrome de Down. En las dos primeras, en las que la patogenia se refiere a herencia autosómica recesiva, por razones culturales y educativas, la frecuencia es muy alta en la población indígena.
Estos pacientes tienen menor acceso a los sistemas de salud debido a su aislamiento de los centros de poder del país. Por ello reciben un manejo inadecuado y su calidad de vida es menor al que debiera esperarse en otras circunstancias.
Como también podemos observar en el trabajo previo, hay muchos avances en el manejo actual de estas patologías. Nuestra propuesta se encamina a colaborar para que estos pacientes y sus familias tengan una mejor calidad de vida, a la que como panameños tienen derecho.
Esta fundación estará compuesta por profesionales de la medicina involucrados con las genodermatosis y empresarios privados, todos ellos con el único propósito de brindar un cuidado médico correcto y apoyo familiar, con el fin de que el porcentaje de sobre vivencia y la calidad de vida de muchos pacientes puedan ser mejorados; sustentados en cuatro áreas estratégicas: la investigación, la capacitación, la generación y difusión de información y la consulta médica integral.
La definición de las áreas estratégicas, se fundamenta en la siguiente definición del problema que se busca atenuar con la fundación. El problema consiste en la falta de información, sobre las genodermatosis y sus posibles tratamientos, lo que repercute en niños y adultos enfermos a los cuales no se les puede aliviar su padecimiento físico y emocional y la falta de información genética, resultado de estudios genéticos y científicos que sirva de base para ofrecer consejo genético a las familias con propensión a la enfermedadActualmente es de más de 100 niños, niñas y algunos adultos que sufren de estas éstas enfermedades en mayor o menor grado, y sus familias cercanas para tener identificadas las personas que tienen probabilidad de ser portadores de la enfermedad. Entre estos se cuentan los tíos y tías maternas, paternas, y los hermanos del enfermo. Hay que insistir que, por lo menos con los pacientes afectados por el síndrome de Kindler, puede haber muchos pacientes no detectados en las montañas de Bocas del Toro, Chiriquí y Veraguas, que se beneficiarían de esta propuesta.
MISION
La fundación es una entidad sin fines de lucro, cuyo propósito es mejorar la calidad de vida de los pacientes con genodermatosis en Panamá. Nos caracterizaremos por la responsabilidad, solidaridad y el deseo de ayudar a los pacientes y sus familias aliviando el sufrimiento que les aqueja. Brindaremos servicios de tipo integral basados en el fortalecimiento de la capacidad de los médicos y otros profesionales de la salud, para lo cual investigamos, difundimos información científica y luchamos por la incorporación y aceptación de los pacientes por la sociedad, así como en el ajuste y adaptación de las condiciones de su entorno. Nuestros servicios están dirigidos prioritariamente a los enfermos y sus familias, al cuerpo médico y a la población en general.
VISION
La fundación velará por la integralidad de los servicios a todo nivel, garantizando la solución a problemas médicos incluyendo urgencias y los asociados a mejorar la calidad de vida de los pacientes, como es el acondicionamiento de las viviendas, el aporte de equipo especial y otros.
VALORES
Desarrollar un sistema de información y divulgación como base para difundir información científica, promover la aceptación e inserción social de los enfermos.
Estrategias:
Promover y apoyar la investigación científica orientada al estudio de las causas y el tratamiento de las Genodermatosis y otras enfermedades severas de la piel.
Estrategia:
Capacitar a los médicos y otros profesionales de la salud en el tratamiento y procedimientos que se deben aplicar por las diferentes especialidades a los enfermos con genodermatosis y otras enfermedades severas de la piel, con miras a constituir equipos especializados de atención integral.
Estrategias:
Brindar Consulta médica integral, con el fin de aliviar el sufrimiento físico y mental de los pacientes con Genodermatosis y otras enfermedades severas de la piel.
Estrategias:
Establecer vínculos formales con fundaciones que velan por fines similares y complementarios, mediante la definición de relaciones de coordinación y la aplicación de procedimientos.
Estrategias:
Fortalecer y consolidar la estructura y procesos organizativos de la fundación, para su sostenibilidad y mejor prestación de servicios
Estrategias
ANALISIS DE FORTALEZAS, OPORTUNIDADES, DEBILIDADES Y AMENAZAS (FODA)
Fortalezas
Recurso humano con el que actualmente cuenta la fundación, Junta y apoyo médico y administrativo, tienen conocimiento y experiencia sobre el ámbito de acción de la Fundación; pero básicamente tienen mística e interés en colaborar.
Se cuenta con apoyo institucional, principalmente del Hospital José Domingo de Obaldía sección pediátrica, para la prestación de sus servicios y del personal médico de esta institución.
La gran voluntad que existe de parte de los miembros fundadores para luchar contra las enfermedades.
La fortaleza mental y moral que demuestran los pacientes con la enfermedad es una gran inspiración para seguir adelante
OportunidadesPaís pequeño, bien comunicado, que permite la efectiva promoción de las acciones de la fundación y realización de campañas para el financiamiento de las mismas.
Relación y buena acogida con medios de comunicación, los cuales se han sensibilizado y comprometido con los propósitos de la fundación.
Contactos internacionales que facilitarán la gestión de recursos.
La existencia de Fundaciones con iguales propósitos en otros países y la posibilidad de integrarse en una red de coordinación y cooperación internacional.
Posibilidades de desarrollar investigación, por la existencia de recursos financieros y materiales en el país y fuera del país.
El contar con la ayuda del Club Activo 20-30 de Panamá.
El apoyo desinteresado de la Clínica de la Piel.
La política social y de salud del país, sobre todo la actitud de la Primera Dama actualmente.
DebilidadesFalta de recurso económico propio para la operación continúa de la fundación.
Desorden en procedimientos administrativos y operativos
Ausencia de reuniones periódicas para la toma de decisiones
Intromisión de opiniones no especializadas en el campo médico para establecer acciones acerca de los objetivos de la Fundación.
Falta de un local propio
Amenazas
Ausencia de claridad en los padres de familia acerca de la exclusividad de asuntos médicos como prioritario de la labor de la fundación.
Posibilidad de que se privatice la medicina.
La existencia de tantas fundaciones que lucha cada uno por obtener fondos públicos y privados, todos para una buena causa, lo que puede provocar un desgaste en el público y la eventual decisión de no colaborar más.
| ESTRATEGIA | ACTIVIDAD PRIORITARIA |
ACTORES INVOLUCRADOS | RESPONSABLE | TIEMPO | |||
| 1 | 2 | 3 | 4 | ||||
| 1.1 Brindar información genética con el fin de prevenir el nacimiento de niños enfermos. | 1.1.1 Constitución de una base de datos con los resultados del objetivo No.2, que será parte del sistema de información general. | ||||||
| 1.1.2 Diseño de un mecanismo de transferencia de los registros de la base de datos. | |||||||
| 1.2 Brindar información a médicos y otros profesionales de la salud para una mejor atención integral de los pacientes. | 1.2.1 Diseño y puesta en operación de un sistema de información con información actual y novedosa sobre investigaciones, nuevos tratamientos otros | ||||||
| 1.3 Divulgar al público en general información sobre la enfermedad y las características y calidades de los pacientes, con el fin de promover su inserción y aceptación en la sociedad. | 1.3.1 Contar con un registro de la población beneficiaria, detallando características del paciente, condiciones y potencialidad. |
||||||
OBJETIVO ESTRATEGICO No.2
Promover y apoyar la investigación científica orientada al estudio de las causas y el tratamiento de las GENODERMATOSIS y otras enfermedades severas de la piel.
| ESTRATEGIA | ACTIVIDAD PRIORITARIA |
ACTORES INVOLUCRADOS | RESPONSABLE | TIEMPO | |||
| 1 | 2 | 3 | 4 | ||||
| 2.1 Realizar procesos de investigación, que permitan la clasificación de cada uno de los pacientes y su enfermedad, resultado de estudios especializados y como base para un adecuado diagnóstico y definición del tratamiento. | 2.1.1 Elaboración de estudios especializados por paciente | ||||||
| 2.1.2 Clasificación de los pacientes | |||||||
| 2.1.3 Diagnóstico y definición de tratamientos | |||||||
OBJETIVO ESTRATEGICO No.3
Capacitar a los médicos y otros profesionales de la salud en el tratamiento y procedimientos que se deben aplicar por las diferentes especialidades a los pacientes con GENODERMATOSIS y otras enfermedades severas de la piel, con miras a constituir equipos especializados de atención integral.
| ESTRATEGIA | ACTIVIDAD PRIORITARIA |
ACTORES INVOLUCRADOS | RESPONSABLE | TIEMPO | |||
| 1 | 2 | 3 | 4 | ||||
| 3.1 Constituir equipos de atención integral especializados, capacitando médicos, enfermeras y otros servidores de la salud, de todas las especialidades, sobre el mejor tratamiento y las innovaciones para tratar las G y otras enfermedades severas de la piel. | 3.1.1 Identificar las principales necesidades de capacitación. | ||||||
| 3.1.2 Identificar las principales fuentes de capacitación. | |||||||
| 3.1.3 Promover procesos de capacitación | |||||||
OBJETIVO ESTRATEGICO No.4
Brindar Consulta médica integral, con el fin de aliviar el sufrimiento físico y mental de los pacientes con GENODERMATOSIS y otras enfermedades severas de la piel.
| ESTRATEGIA | ACTIVIDAD PRIORITARIA |
ACTORES INVOLUCRADOS | RESPONSABLE | TIEMPO | |||
| 1 | 2 | 3 | 4 | ||||
4.1 Potenciar el uso de los recursos institucionales disponibles y así maximizar su aprovechamiento y su integración. |
4.1.1 Construir una base de datos sobre las diferentes entidades relacionadas con las actividades de la fundación. Definiendo la oportunidad que representa para esta. | ||||||
| 4.1.2 Establecer relaciones formales con estas entidades, convenios, acuerdos, cartas de entendimiento. | |||||||
4.2 Fortalecer la capacidad de atención del Hospital de Niños, promoviendo la constitución de una Unidad especializada en G y otras enfermedades severas de la piel. |
4.2.1 Involucrar al personal del HN en la fundación | ||||||
| 4.2.2 Establecer relaciones formales con el HN, mediante la coordinación de acciones y la definición de procedimientos | |||||||
| 4.2.3 Orientar los resultados de los objetivos 1, 2 y 3 al personal del HN | |||||||
| 4.2.4 Constituir la Unidad de Servicios especializados | |||||||
| 4.3 Brindar consulta medica con recursos de la fundación e integrando recursos de otras entidades. | 4.3.1 Contar con un registro de pacientes y sus necesidades |
||||||
| 4.3.2 Identificar fuentes de servicios ó recursos en función de la actividad 4.1.1 | |||||||
| 4.3.3 Definir tratamientos integrales por paciente, según sus condiciones. | |||||||
OBJETIVO ESTRATEGICO No.5
Establecer vínculos formales con fundaciones que velan por fines similares y complementarios, mediante la definición de relaciones de coordinación y la aplicación de procedimientos.
| ESTRATEGIA | ACTIVIDAD PRIORITARIA |
ACTORES INVOLUCRADOS | RESPONSABLE | TIEMPO | |||
| 1 | 2 | 3 | 4 | ||||
5.1 Promover la constitución de una red internacional de fundaciones con fines similares |
5.1.1 Identificar organizaciones afines y similares | ||||||
| 5.1.2 Promover la constitución de la red | |||||||
| 5.1.3 Constituir la Red | |||||||
5.2 Desarrollar relaciones de coordinación con las Fundaciones afines en Panamá en un ambiente de complementariedad. |
5.2.1 Diferenciar roles e intereses | ||||||
| 5.2.2 Definir mecanismos de coordinación | |||||||
| 5.2.3 Definir procedimientos para trabajo en conjunto | |||||||
| 5.2.4 Establecer relaciones formales de trabajo en conjunto | |||||||
| ESTRATEGIA | ACTIVIDAD PRIORITARIA |
ACTORES INVOLUCRADOS | RESPONSABLE | TIEMPO | |||
| 1 | 2 | 3 | 4 | ||||
Puesta en operación de la estructura organizativa propuesta, |
Gestión del Recurso Humano | ||||||
| Inducción del recurso Humano | |||||||
| Operatividad de la Junta Directiva, incluye inducción y aprobación de procedimientos | |||||||
Aplicación de manuales de procedimientos, de control interno y operativo. |
Revisión y ajuste de procedimientos | ||||||
| Aplicación de los procedimientos | |||||||
Gestión de recursos para el financiamiento de la fundación |
Realizar un programa de aporte por parte de los socios | ||||||
| Realizar una campaña de aporte por parte de personas, empresas e instituciones interesadas en los fines de la fundación | |||||||
| Formular proyectos y propuestas específicas para el financiamiento de los objetivos 1, 2, 3 y 4. | |||||||
La propuesta se hace con fundamento en la situación actual de la organización, las definidas como áreas estratégicas y sus objetivos.
Se propone una estructura básica, dinámica y flexible, que deberá modificarse conforme se ejecuten acciones y se logren los objetivos operativos y estratégicos de la fundación.
Como se puede ver en el organigrama, entre las áreas funcionales se propone una dinámica de cogestión, esto porque los objetivos de cada una de estas áreas, se deben tratar en forma integral, considerando cada una de ellas la dependencia y relaciones de coordinación que se deben establecer para el logro de los fines de la fundación.
Para cada una de estas áreas funcionales se desarrollará una subestructura propia, en función de las propuestas y proyectos que se implementen, las cuales pueden considerar acciones para varias áreas a la vez.
Como componentes fijos se definen la Junta Administrativa, la Dirección Ejecutiva y el apoyo administrativo; también se consideran componentes temporales como servicios especiales para apoyo en la formulación de propuestas y proyectos, para los servicios contables, u otros que se consideren necesarios.
Fundacion
Brinda orientaciones estratégicas y supervisa que la operación de los proyectos, reflejen los objetivos y políticas de la Fundación. Por lo que se puede definir como el órgano de dirección y conducción estratégica de la fundación.
Las principales funciones serán:
Las principales funciones son:
Principales funciones:
Principales funciones:
Coordinaor De Proyectos Por Area Funcional
Principales funciones:
GRUPOS DE APOYO EN GENODERMATOSIS
EPIDERMOLISIS BULOSA
Dystrophic Epidermolysis Bulosa Reserach Association of America (DEBRA)
www.debra.org
5West 36th Street, suite 404
Ney York, NY 10018
(212) 868 1573; fax (212) 868 9296
Director Ejecutivo: Andrew McCluskey
Tiene filiales en Chile, México y Costa Rica.
ICHTHYOSIS
Foundation for Ichthyosis and Related Skin Types (FIRST)
www.saclyskin.org
650N. Cannon Avenue, Suite 17
Lansdale, PA 19446
(800)545-2386 ; FAX (215) 631 1413
Director Ejecutivo : Jean Pickford.
NEUROFIBROMATOSIS I
National Neurofibromatosis Foundation
www.nf.org
95Pine Street, 16th floor
New York, NY 10005
(800) 323 7938
FAX: (212) 747 0004
Director Ejecutivo Peter Bellerman, President.
XERODERMA PIGMENTOSUM
Xeroderma Pigmentosum Registry
University of Medicine and Dentristy of New Jersey
Department of Pathology
Med Sci Bldg Rm C-520
185 South Orange Avenue
Newark, NJ 07103-2714
(201) 982-4405
Director Ejecutivo: W. Clark Lambert, MD, Ph.D.
ALBINISMO
National Organization for albinism and Hypopigmentation
www.albinism.org
P.O.Box 959
East Hamstead, NH 03826-0959
(800) 472 2310
FAX (603) 887 6049
Director Ejecutivo Michael McGowan, President.
"GRANDES EDUCADORES HAN SABIDO SIEMPRE QUE EL APRENDIZAJE NO ES ALGO QUE ESTÁ LIMITADO AL SALÓN DE CLASES O QUE FORZOSAMENTE DEBE SER EMPRENDIDO BAJO LA SUPERVISIÓN DE LOS MAESTROS."
BILL GATES

"EXISTIR NO SIGNIFICA ACEPTAR LO QUE UNO ES; SIGNIFICA CREAR OTRO YO QUE NO EXISTE."
J. CHATEAU